An open-source Python package for accessing and analyzing NMR data from either custom input or NMR-STAR files from the BMRB.
pip install makeshift-nmrimport makeshift as ms
# Fetch and parse a BMRB entry into tidy chemical shifts
cs = ms.ChemicalShifts.from_bmrb(5363)
cs.data # one row per shift: Seq_ID, Comp_ID, Atom_ID, Atom_type, Val
cs.sequences() # one row per entity: ID, polymer type, sequence
# Re-reference shifts
cs = ms.ChemicalShifts.from_bmrb(4527, reref="lacs", calc_csi=True)
cs.reref_offsets # {atom: offset applied}
# Build an assigned peak list (e.g. for an HSQC)
peaks = cs.peaklist()
peaks.data| Module | What it does |
|---|---|
makeshift (core) |
ChemicalShifts, NMRStarEntry, PeakList — fetch/parse BMRB entries, extract shifts, sequences, relaxation/order-parameter data, build peak lists. |
makeshift.reref |
LACS and PANAV chemical-shift re-referencing (via ChemicalShifts.reref). |
makeshift.spectra |
Read Sparky .ucsf spectra (Spectrum), pick peaks, and align peak lists (map_peaklists). |
makeshift.relaxation |
CPMG dispersion pipeline (CPMGExperiment) and RelaxationProfile — RelaxDB-style per-residue dynamics from deposited R1/R2/NOE. |
makeshift.hydronmr |
Predict per-residue T1/T2/NOE from a PDB structure (run). |
makeshift.talosn |
Predict backbone torsion angles, S2 order parameters, and secondary structure from chemical shifts via the NIH TALOS-N binary (TalosN). |
makeshift.utils |
Dependency-light helpers: dataset/structure fetching (fetch_structure), constants. |
See demos/ for worked examples:
quick_start.ipynb(core workflow),reref.ipynb(re-referencing),cpmg_demo.ipynb(the CPMG pipeline),bmrb_relaxation_demo.ipynb(deposited relaxation → dynamics profile)talosn_demo.ipynb(TALOS-N prediction).
BMRB shifts are sometimes mis-referenced — a constant offset shifts every peak
of a given nucleus. ChemicalShifts.reref corrects this in place using one of
two methods:
- PANAV (Wang & Wishart 2005) — uses rarely-misreferenced HA shifts to assign secondary structure, then aligns N/CA/CB to curated per-structure reference distributions (Wang & Jardetzky 2002).
- LACS (Wang & Markley 2009) — fits secondary shift vs. CSI so the random-coil regime intercepts at the origin; covers CA, CB, C′, N, and HN.
cs = ms.ChemicalShifts.from_bmrb(4527)
cs.reref(method="panav") # or "lacs"
print(cs.reref_offsets) # {'N': ..., 'CA': ..., 'CB': ..., ...}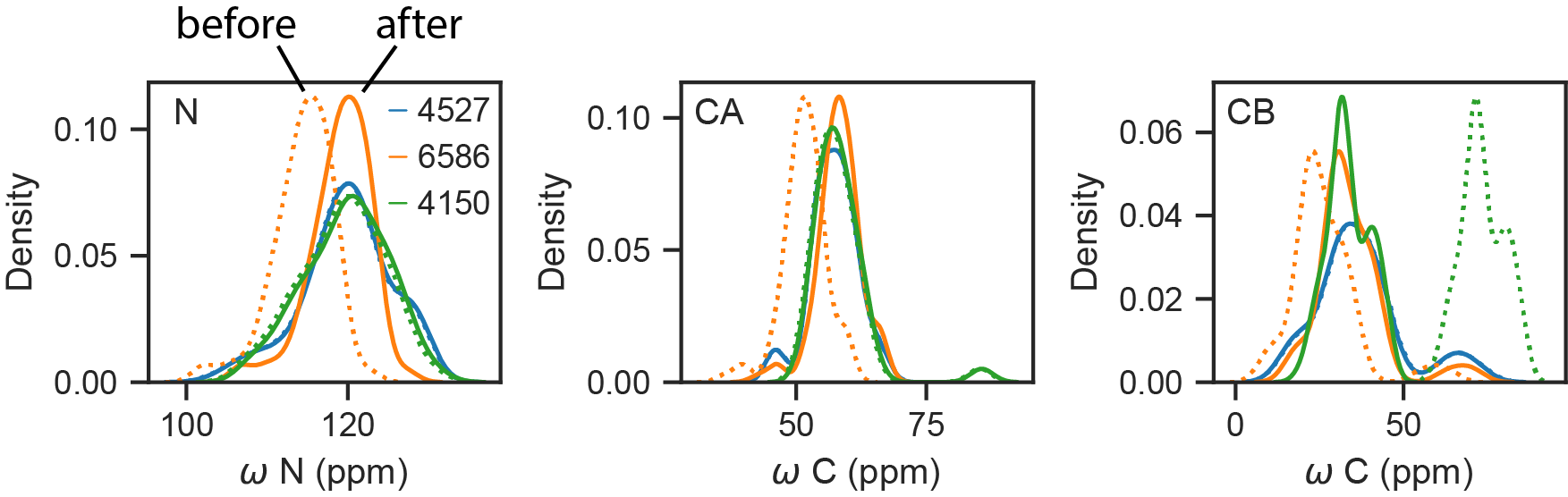
Entry 4527 is correctly referenced; entries 6586 and 4150 have been described in the literature as needing re-referencing. The two methods have not yet been extensively compared.
NMRStarEntry extracts any deposited relaxation data, and RelaxationProfile
turns it into a per-residue dynamics analysis in the style of RelaxDB
(Wayment-Steele, El Nesr et al.).
Pull deposited data straight from an entry:
entry = ms.NMRStarEntry.from_bmrb(25013)
entry.datasets() # which data types the entry holds
entry.relaxation("T2") # R2 (also "T1"/"R1", "T1rho", "NOE") — units-aware
entry.order_parameters() # model-free S2 (S2, Tau_e, Rex)
entry.data_loop("spectral_density_values", "_Spectral_density") # anything elseRelaxationProfile assembles R1/R2/NOE into the R₂/R₁ observable, compares it to
a HYDRONMR rigid-body prediction, and labels each residue by motional regime:
from makeshift.relaxation import RelaxationProfile
prof = RelaxationProfile.from_bmrb(25013) # pulls T1/T2/NOE, aligns to the sequence
prof.add_rigid_prediction() # structure: deposited PDB → RCSB, else AlphaFold → AFDB
print(prof.label()) # per-residue motion string
prof.plot("R2_R1")The structure for the rigid prediction can be a local PDB, a PDB id (fetched
from RCSB), or a UniProt accession (fetched from AlphaFold DB) — e.g.
add_rigid_prediction("1WRP"), ("P0DP23"), or ("model.pdb"); with no
argument it uses the entry's own cited PDB or AlphaFold model. makeshift does not
predict structure itself.
Label tokens: A ordered, ^ µs–ms exchange (elevated R₂/R₁), v ps–ns motion
(hetNOE ≤ 0.65), b both, . peak missing, t disordered terminus, p proline.
makeshift.talosn wraps the NIH
TALOS-N binary (Shen &
Bax, J. Biomol. NMR 2013), which predicts backbone φ/ψ torsion angles,
per-residue S2 order parameters, and secondary structure from assigned backbone
chemical shifts using a trained neural network.
The binary and its database aren't bundled — they're downloaded on demand from
NIH (under their Terms of Use,
which the installer prints) into a data_dir you choose. Keep that path in a
variable and pass the same one to install and to each TalosN:
from pathlib import Path
from makeshift import talosn
data_dir = Path.home() / "talosn_data"
talosn.install_talosn_data(data_dir=data_dir) # one-time, ~ a few hundred MB
tn = talosn.TalosN.from_bmrb(4527, data_dir=data_dir)
tn.run() # or run(auto_install=True) to fetch the binary on first use
tn.order_parameters # predS2.tab — per-residue S2
tn.torsion_angles # pred.tab — φ/ψ per residue + confidence class
tn.secondary_structure # predSS.tab — helix/sheet/coildata_dir defaults to inside the installed package if omitted (usually not what
you want for a few-hundred-MB download).
NMR-STAR files are organised around saveframes, each belonging to a category
(e.g. assigned_chemical_shifts, entity, sample). The three you interact
with most:
- Entry — a single BMRB deposition (one
.strfile). - Entity — a distinct molecular species (protein, DNA strand, ligand), each
with its own
Entity_ID. - Chemical shift list — the
_Atom_chem_shiftloop inside anassigned_chemical_shiftssaveframe; one row per observed shift.
MIT License.
makeshift.talosn downloads and runs the TALOS-N binary, which is distributed
separately by NIH under its own
Terms of Use (including no
redistribution without permission from the authors); those terms govern the
downloaded software, not this wrapper.
- The Biological Magnetic Resonance Bank (BMRB) for maintaining and sharing NMR data.
- The Bax lab at NIH for TALOS-N.